|
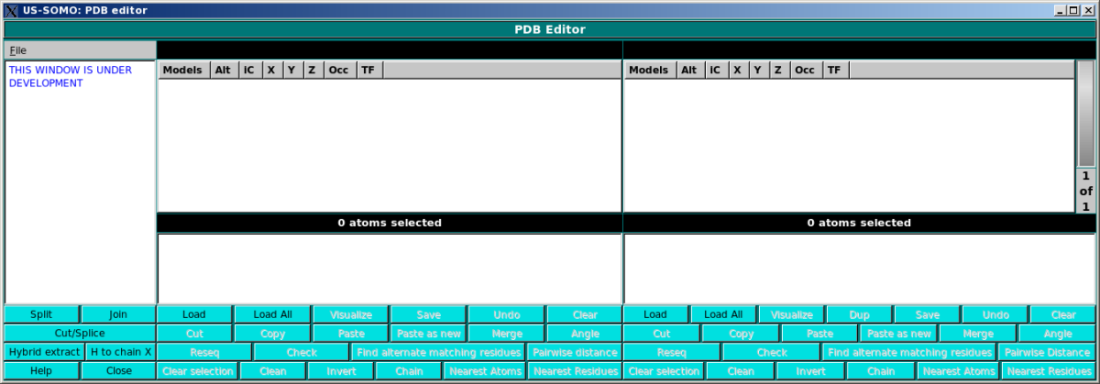
In the main panel, the user will be able visualize in detail PDB files content, and
perform multiple operations on them. The screen is divided in two identical panels,
so that content can be copied/edited/cut/pasted between two different files.
The right panel supports multiple files and has a wheel which allows scrolling through the loaded files.
-
Load. This function loads a PDB file. Notice: It will only load the first model
of a multi-model NMR style PDB file. Use Load All to load all models of an NMR style PDB file.
-
Load All. This function load a PDB file. All models of a NMR style PDB file will
be loaded.
-
Visualize. This function launches a RASMOL window showing the current PDB file.
-
Dup. This function is only present in the right panel. It makes a new Copy of
the current structure in the center panel and Paste as new the copy onto the right panel.
-
Save. This function saves the current PDB file.
-
Undo. This function undoes the last operation.
-
Clear This function clears the panel of files.
-
Cut This function cuts the currently selected models/chains/residues/atoms.
-
Copy This function copies the currently selected models/chains/residues/atoms into a copy buffer.
-
Paste This function pastes the copy buffer onto the current structure.
-
Paste as new This function pastes the copy buffer as a new structure.
-
Merge This function is under development. It allows merging of two PDB files.
-
Angle This function is enabled when exactly three atoms are selected. The angles
are reported in the leftmost panel for each of the three possible angles.
-
Reseq This function is under development. It will resequence the atom and residue
nubmers of a PDB.
-
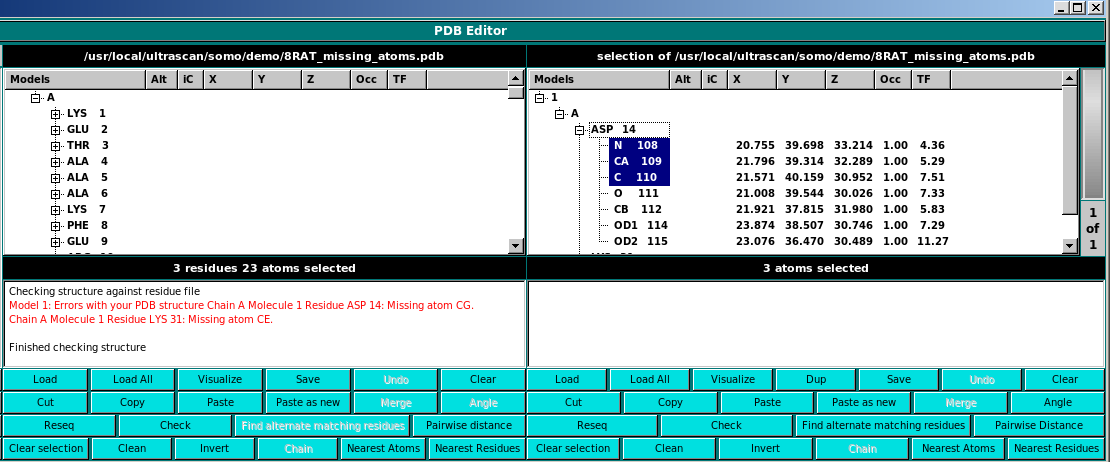
Check This function checks the current structure for residue matching errors.
Note: it currently may report problems even when none exist. Check the textbox in the panel
for details. If it says the model is ok, it is ok. You can optionally select residues for
errors. Then, it is convenient to Copy and then in the other panel Paste as new
to further investigate the issues.
-
Find alternate matching residues If exactly one residue is selected, this function
is enabled and will check the residue table for alternative residue names. This is useful
if you have a miscoded residue. You can replace the residue name with the correct one and Save it.
-
Pairwise Distance This function computes minimum pairwise distances between selected atoms.
A dialog is presented asking the minimum number of pairwise distances to find (default 1).
A second dialog is presented asking to restrict the search to specific atoms. Press CANCEL to disable
this restriction. Then a report is presented in the left most text panel listing the N minimum
pairwise distances. The format is Model~Chain~Residue~Atom~Model~Chain~Residue~Atom distance.
-
Clear selection This clears all selections in the panel.
-
Clean Sometimes selections can get a bit odd. For example you can
arbitrary models, chains, residue and atoms selected by manual interaction with the
PDB structure tree. Cleaning the selection will make this consistent.
A summary of selections is always displayed below the PDB tree view.
-
Invert This function inverts the current selection. I.e. everything selected
will become unselected and vice-versa.
-
Nearest Atoms This function finds the N nearest atoms to the current selection and
select them. A dialog box shows up requesting the number of atoms to find. After selection,
the selection may not be visible. A convenient way to see them is to immediately Copy
and Paste as new. You may still have to open residues in the tree to see individual
atoms.
-
Nearest Residues This function finds the N nearest residues to the current selection.
|