
| |
Manual |
The calculation of the fractal dimension of macromolecules from their atomic structure is a new feature available from the July 2024 US-SOMO release. It was implemented during an investigation into the reason behind the relatively small differences in computing the hydrodynamic parameters between the GRPY and ZENO methods (see the "History" section in the main US-SOMO Help), but it could find other applications, and for this reason it is offered to the general user.
The first section of this panel present the general Fractal Dimension options:
 |
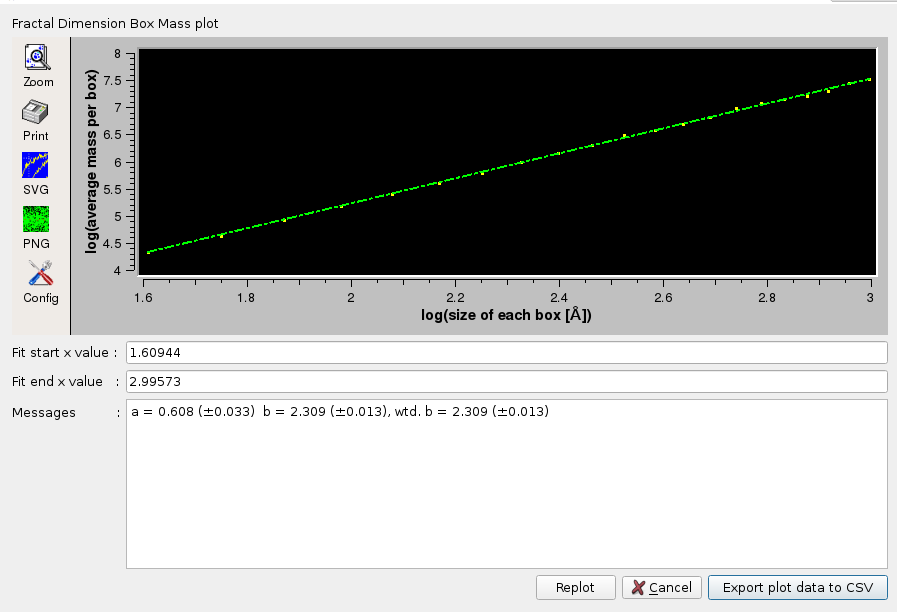
The first method is a general box counting algorithm that returns the mass fractal dimension Dm, as decribed in Liebovitch and Toth, "A fast algorithm to determine fractal dimensions by box counting.", Phys. Lett. A, 141:386-390, 1989. To compute the fractal dimension via Box Mass, the program iterates Number of Steps times from Start [Å] to End [Å]. For each step, a grid of boxes is placed over the protein. The average mass of each box containing at least one atom of the protein is computed. The logarithm of the average mass per box versus the logarithm of the edge size of each box is collected (and optionally plotted). The slope of this line, determined through linear regression, provides an estimate of the fractal dimension.
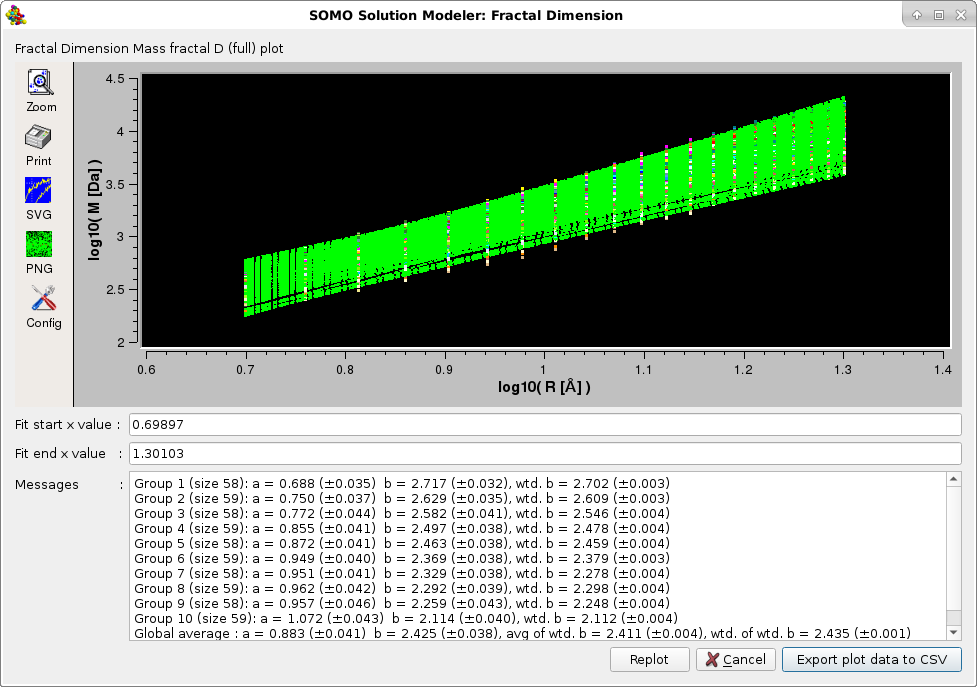
The second and third options also provide the mass fractal dimension Dm, following the work of Enright and Leitner, ("Mass fractal dimension and the compactness of proteins", Phys. Rev. E 71, 011912-1/9, 2005), where the structure is divided in "slices" selecting the nearest CA atoms within 10%, 10-20 %, 20-30 %, etc. from the center of mass of the protein. Each set is used as centers of concentric spheres or radius R containing atoms of total mass M, and Dm is computed as the slope of the plot of log10 M vs log10 R. Dm can be evaluated from a particular "slice" (Mass Fractal D (slice) option), or as an average between all slices (Mass Fractal D (full) option). See the Enright and Leitner paper for a full description of this method. In our implementation, each linear fit of of log10 M vs log10 R produces data with SD of the intercept and slope, and Dm is calculated as the unweigthed and weighted averages (± their SD) within each slice. For the full version, an unweighted and a weighted average are calculated from the the weighted averages (± their SD). Weighting reduces the influence of poor linear regression fittings in the final results.
The third method returns the surface fractal dimension Ds using the dependence of the accessible surface area of the structure as a function of the probe radius r, using the same rolling sphere algorithm ASAB1 as described here. Since ASA ~ r2 - Ds, the slope of a double log10 plot of ASA vs. r will enable the determination of Ds. See T.G Dewey, Fractals in molecular biophysics, Chapter 2 "Fractal Aspects of Protein Structure". Oxford University Press, Oxford, New York, 1997; and references therein.
Only in Expert mode, the Rg/Dm or Rg/Ds ratios are also reported for the chosen calculations.
 |
 |
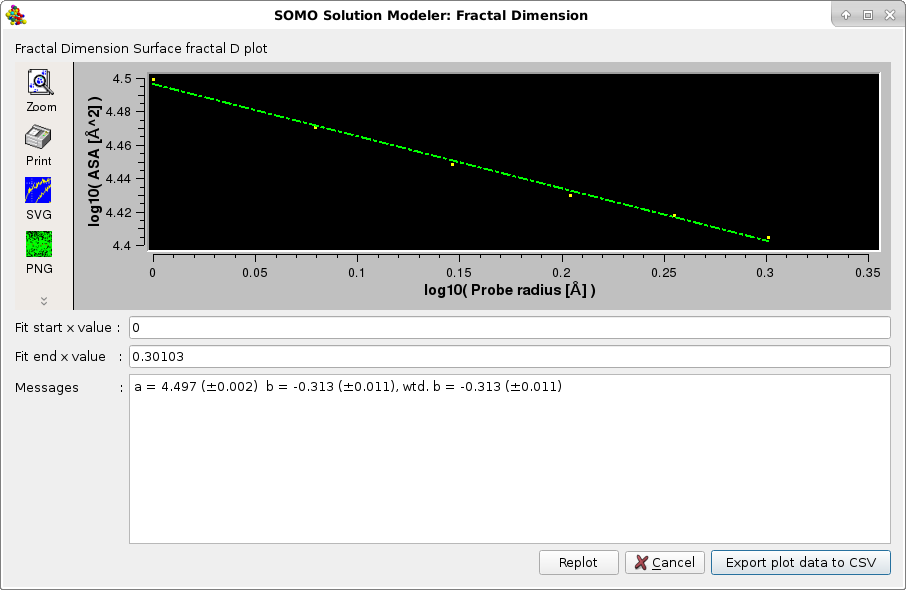 |
The plots are interactive, the x-axis starting and ending values can be changed and the fitting redone by pressing the Replot button. The final plots data can be saved in a csv-style file. The Default setting for this option is disabled.
In the second section, some common parameters for the Box Mass and Mass fractal D methods are set:
 |
 |
 |
This document is part of the UltraScan Software Documentation
distribution.
Copyright © notice.
The latest version of this document can always be found at:
Last modified on June 13, 2024